Analyze a dataset in memory#
Here, we’ll analyze the growing dataset by loading it into memory.
This is only possible if it’s not too large.
If your data is large, you’ll likely want to iterate over the dataset to train a model, the topic of the next page ().
import lamindb as ln
import lnschema_bionty as lb
import anndata as ad
💡 lamindb instance: testuser1/test-scrna
ln.track()
💡 notebook imports: anndata==0.9.2 lamindb==0.61.0 lnschema_bionty==0.35.1 scanpy==1.9.6
💡 saved: Transform(uid='mfWKm8OtAzp8z8', name='Analyze a dataset in memory', short_name='scrna4', version='0', type=notebook, updated_at=2023-11-20 22:27:22 UTC, created_by_id=1)
💡 saved: Run(uid='OWJnVERnirYa8yS9kYFS', run_at=2023-11-20 22:27:22 UTC, transform_id=4, created_by_id=1)
ln.Dataset.filter().df()
| uid | name | description | version | hash | reference | reference_type | transform_id | run_id | file_id | storage_id | initial_version_id | visibility | updated_at | created_by_id | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| id | |||||||||||||||
| 1 | pscxqpvwQa4OPAZUyTXQ | My versioned scRNA-seq dataset | None | 1 | 9sXda5E7BYiVoDOQkTC0KB | None | None | 1 | 1 | 1.0 | None | NaN | 0 | 2023-11-20 22:26:39.943506+00:00 | 1 |
| 2 | xDJG50qUj6fnqYEgYaA5 | My versioned scRNA-seq dataset | None | 2 | -J1PZEjWCBP0OptD6HtZ | None | None | 2 | 2 | NaN | None | 1.0 | 0 | 2023-11-20 22:27:09.681845+00:00 | 1 |
dataset = ln.Dataset.filter(name="My versioned scRNA-seq dataset", version="2").one()
dataset.files.df()
| uid | storage_id | key | suffix | accessor | description | version | size | hash | hash_type | transform_id | run_id | initial_version_id | visibility | key_is_virtual | updated_at | created_by_id | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| id | |||||||||||||||||
| 1 | pscxqpvwQa4OPAZUyTXQ | 1 | scrna/conde22.h5ad | .h5ad | AnnData | Human immune cells from Conde22 | None | 57612943 | 9sXda5E7BYiVoDOQkTC0KB | sha1-fl | 1 | 1 | None | 0 | True | 2023-11-20 22:26:39.939281+00:00 | 1 |
| 2 | z0DtvXhdZMNVoqIagoG1 | 1 | None | .h5ad | AnnData | 10x reference adata | None | 857752 | SAuVZAKKM_Ypj_0SdrhDIg | md5 | 2 | 2 | None | 0 | True | 2023-11-20 22:27:07.563593+00:00 | 1 |
If the dataset doesn’t consist of too many files, we can now load it into memory.
Under-the-hood, the AnnData objects are concatenated during loading.
The amount of time this takes depends on a variety of factors.
If it occurs often, one might consider storing a concatenated version of the dataset, rather than the individual pieces.
adata = dataset.load()
The default is an outer join during concatenation as in pandas:
adata
AnnData object with n_obs × n_vars = 1718 × 36508
obs: 'donor', 'tissue', 'cell_type', 'assay', 'n_genes', 'percent_mito', 'louvain', 'file_uid'
obsm: 'X_umap', 'X_pca'
The AnnData has the reference to the individual files in the .obs annotations:
adata.obs.file_uid.cat.categories
Index(['pscxqpvwQa4OPAZUyTXQ', 'z0DtvXhdZMNVoqIagoG1'], dtype='object')
We can easily obtain ensemble IDs for gene symbols using the look up object:
genes = lb.Gene.lookup(field="symbol")
genes.itm2b.ensembl_gene_id
'ENSG00000136156'
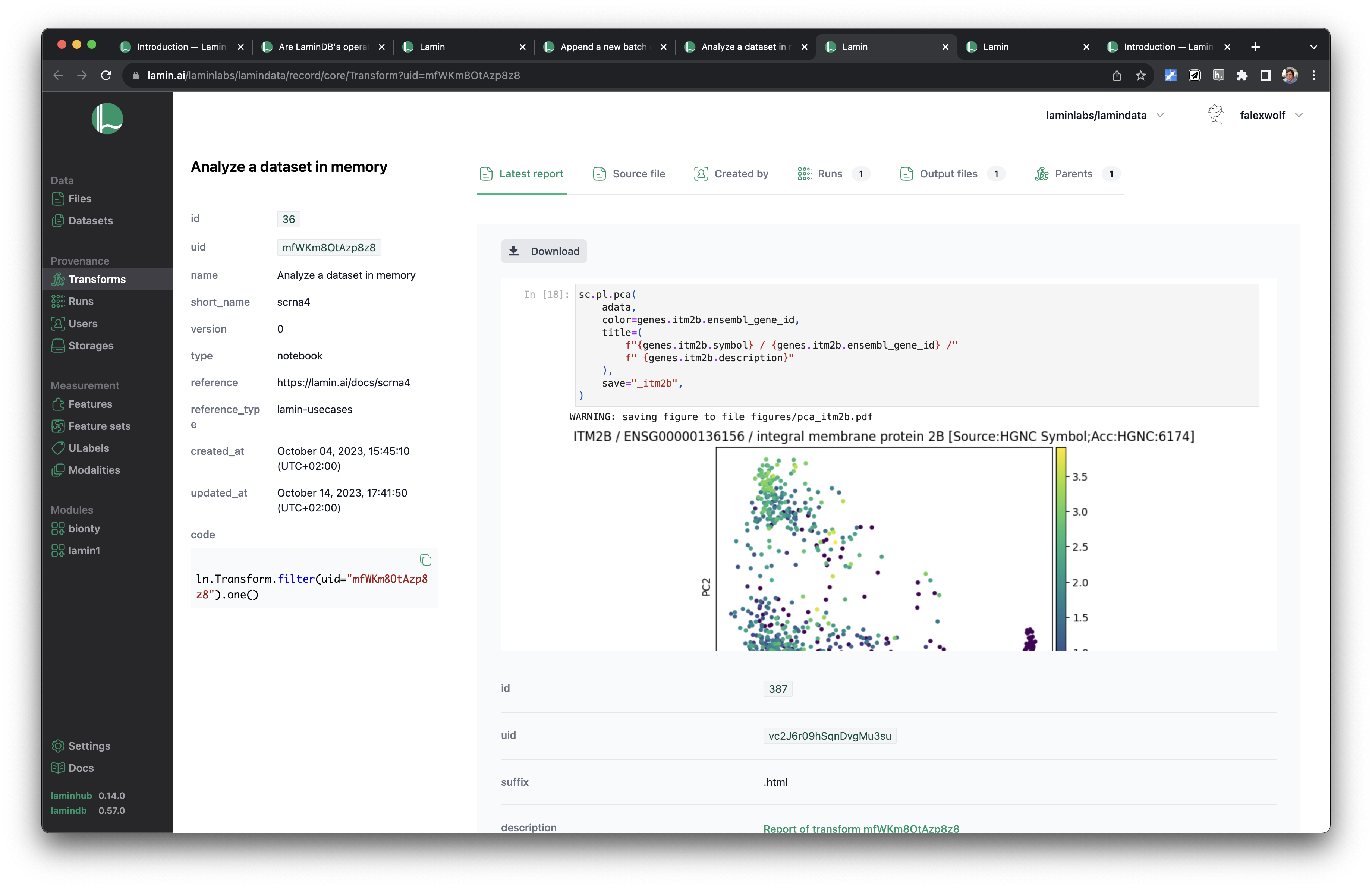
Let us create a plot:
import scanpy as sc
sc.pp.pca(adata, n_comps=2)
2023-11-20 22:27:25,003:INFO - Failed to extract font properties from /usr/share/fonts/truetype/noto/NotoColorEmoji.ttf: In FT2Font: Can not load face (unknown file format; error code 0x2)
2023-11-20 22:27:25,053:INFO - generated new fontManager
sc.pl.pca(
adata,
color=genes.itm2b.ensembl_gene_id,
title=(
f"{genes.itm2b.symbol} / {genes.itm2b.ensembl_gene_id} /"
f" {genes.itm2b.description}"
),
save="_itm2b",
)
WARNING: saving figure to file figures/pca_itm2b.pdf
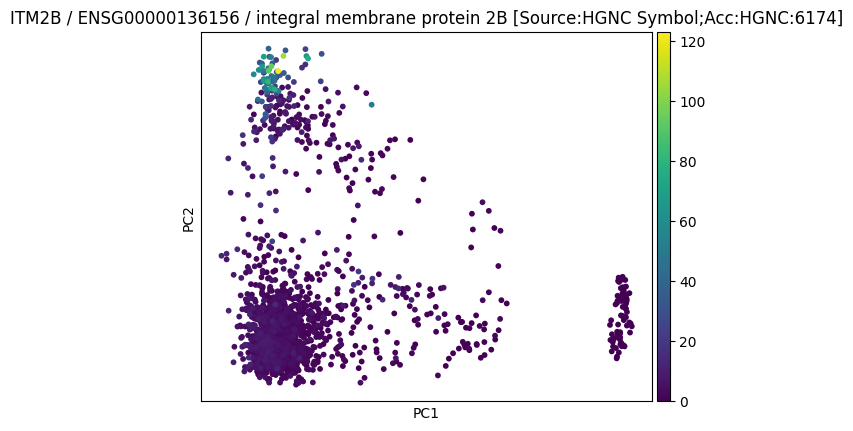
We could save a plot as a pdf and then see it in the flow diagram:
file = ln.File("./figures/pca_itm2b.pdf", description="My result on ITM2B")
file.save()
file.view_flow()
Show code cell output

But given the image is part of the notebook, we can also rely on the report that we create when saving the notebook via the command line via:
lamin save <notebook_path>
To see the current notebook, visit: lamin.ai/laminlabs/lamindata/record/core/Transform?uid=mfWKm8OtAzp8z8